
Protein Language Models: Fluent, but clueless
These models have learned grammar. They have not learned physics.

There’s a beautiful idea at the heart of modern protein AI: that the language of evolution is, at some level, just that - a language. Amino acids are letters. Proteins are sentences. And if you train a large enough model on enough of these sentences, maybe it learns to speak fluently.
This idea is not crazy. It has produced some genuinely remarkable results. But here’s the thing: fluency is not understanding. And in biology, the gap between the two can cost you months of wet-lab time.
At Mandrake, we use autoregressive protein language models (PLMs) every day—ProGen2, ZYMCTRL, and the ecosystem built around them. They are core tools in our protein engineering pipeline, and the whole field is leaning on them. For good reason: they work. But they work within a comfort zone that is narrower than most people realize, and the edges are sharp.
So we stress-tested them. Not on standard benchmarks, but on the questions that actually matter for protein engineering. What we found was a consistent, somewhat unsettling picture that we felt compelled to share.
This is that story.
First: What Even Is a Protein Language Model?
Bear with me for a moment - this will pay off.
A protein is, at its core, a string of amino acids. There are 20 of them (think of them as an alphabet), strung together in chains that can run anywhere from a few dozen to several thousand residues long. This 1D string then folds—through a remarkable process driven by physics and chemistry—into a precise 3D shape. And that shape determines everything: whether a protein catalyses a reaction, binds a drug, edits a genome, or does nothing at all.
For decades, figuring out the relationship between sequence (the string) and function (what it does) was one of biology’s grand unsolved problems. Then, around 2020, researchers noticed something: the statistical structure of protein sequences looks a lot like the statistical structure of text. Both have local patterns (amino acid motifs, like grammatical phrases), long-range dependencies (contacts between distant residues, like subject-verb agreement), and a vast training corpus (hundreds of millions of known protein sequences from nature, i.e. evolution’s 4-billion-year experiment in writing).
So they did the obvious thing: they took the transformer architecture from NLP—the same fundamental engine behind GPT, BERT, and their descendants—and trained it on proteins instead of text.
The results were impressive. Models like ESM-2, ProGen2, and ESM-C learned to assign high probability to sequences that look protein-like, generate novel sequences that fold into real structures, and even predict the effect of mutations on protein stability. ESMFold, built on ESM-2’s representations, can predict a protein’s 3D structure in seconds—a task that once required months of crystallography.
Two broad families emerged:
Autoregressive models (like ProGen2): Read proteins left-to-right, one amino acid at a time. They generate sequences the same way GPT generates text—conditioning each new token on everything that came before. Good for generation. Blind to the future.
Masked language models (like ESM-2): Trained by randomly masking residues and predicting the masked positions from both left and right context. Bidirectional. Better for understanding the whole sequence. Less naturally suited for generation.
Both families are now widely used in protein engineering. Both have been heralded as transformative tools. Both are also, as we’re about to discuss, surprisingly fragile in ways the standard benchmarks completely miss.
These models have learned grammar. They have not learned physics
What we mean by grammar: which amino acids tend to follow which, what motifs are common, how families cluster in sequence space, what an RT domain looks like versus a zinc-finger.
What we mean by physics: how proteins fold into 3D structures, how residues that are far apart in sequence interact when the chain collapses into shape, and how functional constraints—an active site here, a binding interface there—compose into a working machine.
Grammar is impressive. Grammar got us to sequences that look right and fold into plausible shapes. But in protein engineering, “plausible” isn’t enough. You need “functional.” And function lives in physics.
We ran four experiments to probe exactly where the grammar ends and the physics doesn’t begin. Let’s look at what we did and what we found.
Experiment 1: 1D Grammar, Zero 3D Awareness
This is the question at the heart of everything.
When ProGen2 generates a protein one amino acid at a time, left to right, does it have any internal picture of the 3D shape it’s building? Or is it pattern-matching sequences with no awareness of what those sequences do in physical space?
Here’s why this matters. A protein sequence is a 1D string, but the actual object is 3D. Two amino acids that are 250 positions apart in the sequence can be physically touching in the folded protein. These are called long-range 3D contacts, and they are not optional details. They determine whether the protein folds correctly, whether the active site is shaped right, whether the thing actually works.
To test whether ProGen2 sees these contacts, we looked at its attention patterns. In a transformer, attention is the mechanism by which the model decides, at each position, which other positions to look at. If ProGen2 genuinely understands 3D structure, then when it’s generating residue 280, it should be paying special attention to residue 30—because in the folded protein, they’re physically touching, even though they’re 250 positions apart in the sequence.
One subtlety: there are two reasons two residues might attend to each other. First, they could be in structural contact (physically touching in 3D). Second, they could have co-evolved (mutations at one position tend to be compensated by mutations at the other, across evolutionary history). These two signals are correlated but not identical. We specifically wanted the structural signal. So we applied a standard technique called APC (Average Product Correction) to remove the evolutionary background noise. What remains, if anything, should be pure structural awareness.
This is the same principle behind ESMFold: if attention genuinely encodes 3D contacts, you can fold a protein from attention patterns alone. ESM-2 can do this. The question was whether ProGen2 can do anything similar.
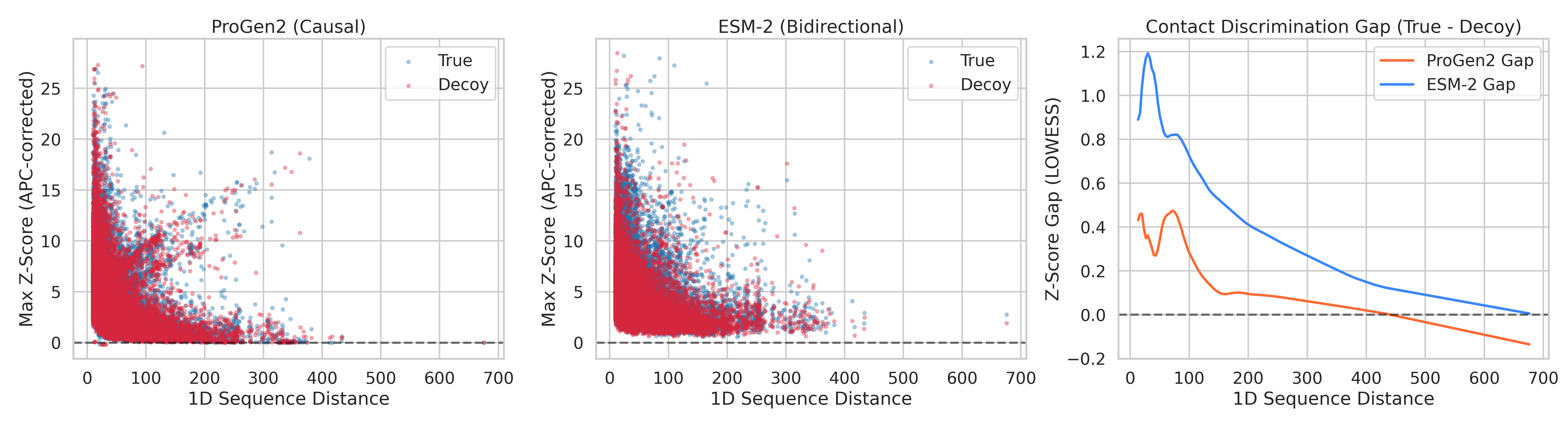
We tested 150 non-redundant protein structures (X-ray resolution ≤ 2.0Å, 100–500 amino acids), across 38,286 contact/decoy pairs. For each protein, we asked: after removing evolutionary background, can ProGen2’s attention distinguish real 3D contacts from decoys at the same sequence distance?
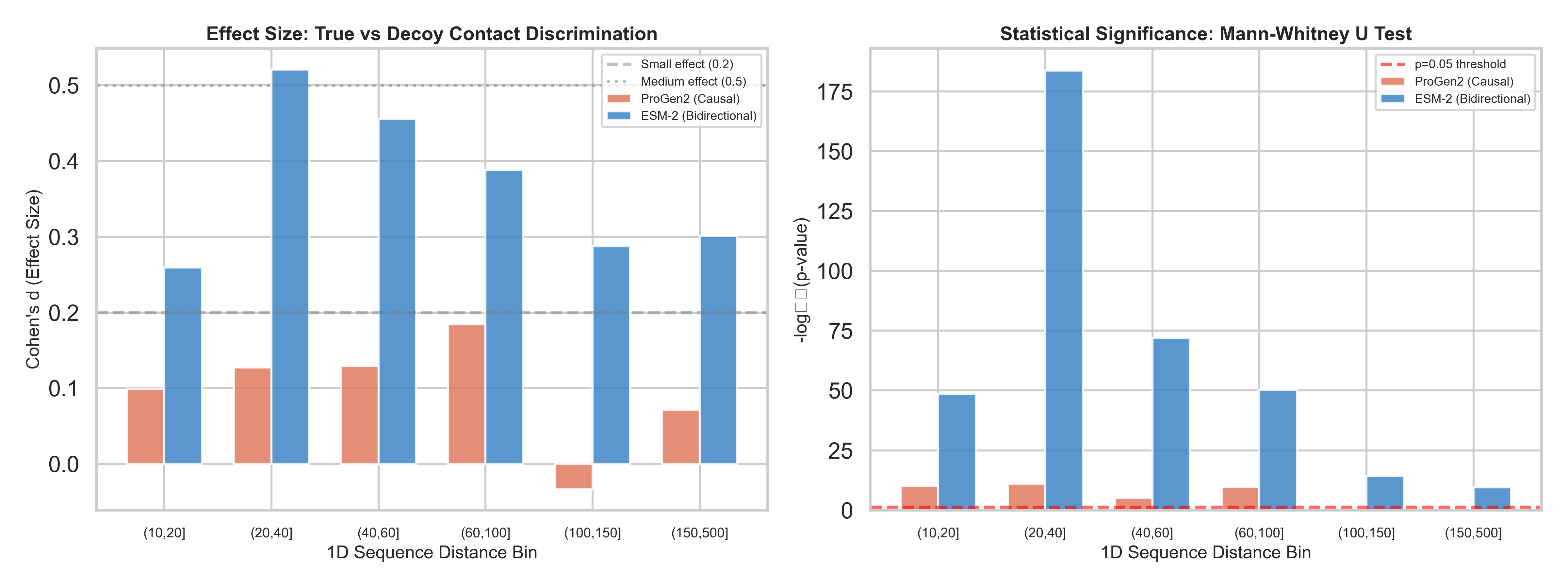
The results across 38,286 contact/decoy pairs:
ProGen2 AUC: 0.527 — barely above coin flip (0.50)
ProGen2 max Cohen’s d: 0.184 — below the threshold for even a “small” effect
Beyond ~100 residues of sequence separation: ProGen2 is statistically indistinguishable from random (p = 0.468 and p = 0.223 for the last two bins)
For comparison, we ran the exact same analysis on ESM-2 (a bidirectional masked model with 12 layers ; matched to ProGen2's depth for a fair architectural comparison):
ESM-2 AUC: 0.611
ESM-2 Cohen’s d peaks at 0.52 — a genuine medium effect
Every single distance bin statistically significant, including the hardest ones (p < 10⁻¹⁰ at 150–500 residues apart)
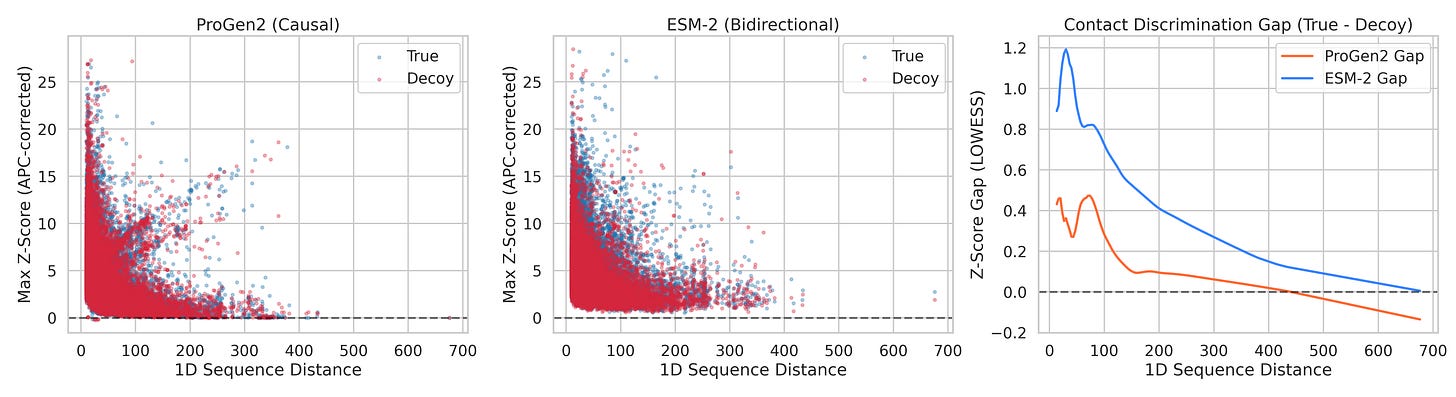
Left: ProGen2 scatter (True vs Decoy Z-scores). Middle: ESM-2 scatter. Right: Z-score gap (True minus Decoy) LOWESS curves overlaid. ProGen2's gap (red) crosses zero around 150 residues and goes negative. ESM-2's gap (blue) stays positive across the entire range
The layer-wise analysis makes the difference even starker. ESM-2 concentrates its structural signal in its final layers—Layer 11 shows Z-score gaps of 1.51, 1.71, and 1.45 across the first three distance bins. It builds a progressive, deep structural representation. ProGen2, despite having 27 layers, never achieves anything comparable.
This doesn't mean ProGen2 learned nothing. It learned excellent 1D grammar ; it knows which amino acids tend to follow which, which motifs are common, what local secondary structure looks like. But 3D contacts, the thing that actually determines whether a protein folds and functions? Essentially invisible to its causal attention.
There’s a paradox buried here worth pausing on. You might think: “Of course - causal attention reads left-to-right, so at any time, it is always working with partial information.” But that’s not actually the explanation.
Look at the short-range bins: at 10-20 residues apart, ProGen2 actually shows some signal. It performs best when it has the least context. The paradox is at long range. When the model is evaluating a contact at position 500, it has already seen 499 residues; nearly the entire protein. It has more than enough context to have built a representation of the protein’s overall architecture. And yet it performs worse than at short range, eventually becoming indistinguishable from random.
The problem isn’t that causal attention sees too little of the protein. The problem is that even when it sees almost all of it, it hasn’t encoded 3D structural relationships into its representations. More context doesn’t help because the model never learned to use context for spatial reasoning in the first place.
Experiment 2: The Copy Bias Trap
This one disturbed us the most.
Take any protein sequence - call it ABC. Now duplicate it: ABCABC. Feed this to ProGen2 and measure how confident the model is at predicting each amino acid.
On the first half (the original), the model behaves normally: uncertain at many positions, per-position perplexity values of 15–25. Expected. Proteins are complex.
On the second half - the repeated copy - perplexity collapses to approximately 1.0. The model predicts every single amino acid with near-perfect confidence. Not because it understands the protein’s structure or function. Because it saw the exact same tokens earlier in the context window and simply copied them.
Here’s the part that should alarm you: this works on random gibberish too. We generated random amino acid strings - no biology, no evolutionary signal, no structural logic - and the collapse is actually worse than for real proteins. It’s pure copy-paste.
This mechanism is the likely culprit behind the high rates of mode collapse we see during candidate generation in our own pipelines.
We then ran a follow-up: give ProGen2 a real protein followed by just the first 25% of a repeat, and let it generate freely. Would it produce a novel protein, or lock into a copy loop?
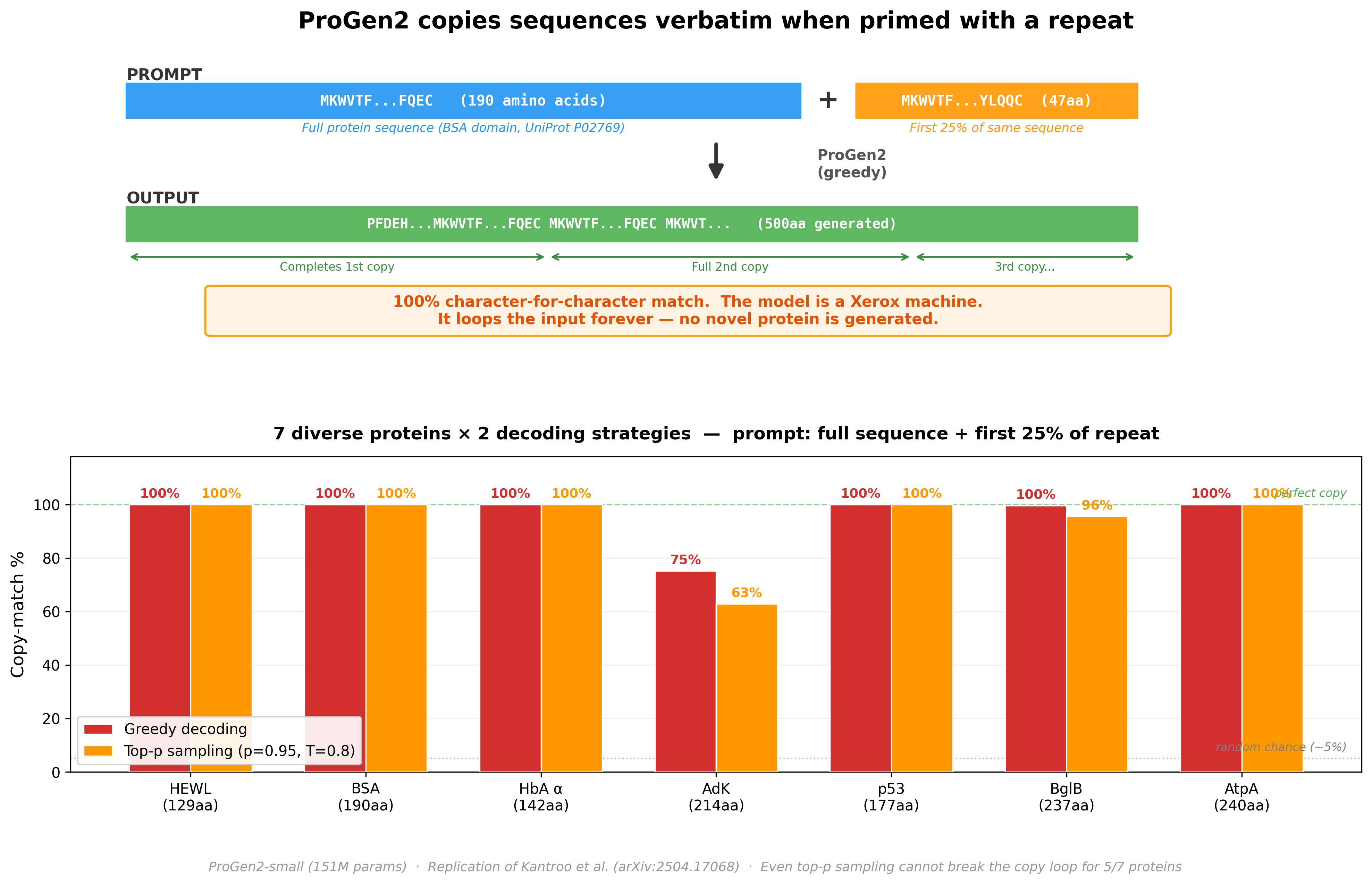
Across 7 diverse proteins (129–240 amino acids): 5 out of 7 produced a 100% character-for-character copy, looping indefinitely. Even with stochastic sampling (top-p = 0.95, temperature = 0.8) - which should introduce randomness - the copy mechanism dominates. The model stops being a protein engineer and becomes a photocopy machine.
This isn’t a ProGen2-specific quirk. Kantroo et al. (arXiv:2504.17068) tested this across ESM2-650M, ESM2-8M, ProGen2-M, CARP-640M, and LC-PLM-1.4B. All auto-regressive transformer-based models show the same collapse. Convolutional architectures (CARP) only collapse for repeats shorter than ~70 residues; BiMamba-based models (LC-PLM) degrade gradually without the catastrophic cliff.
This is an attention-specific architectural vulnerability, and it has a well-understood mechanistic origin. The NLP interpretability literature calls it induction heads - a specific circuit pattern in transformer attention layers that detects and continues repeated token sequences (Olsson et al., 2022).
In text, these circuits make transformers good at in-context learning. In proteins, the same circuits become a liability: they detect any repeated pattern and lock onto it, overriding everything the model knows about biology.
Experiment 3: DMS Variant Scoring
To make the comparison concrete in a practical task, we benchmarked ProGen2-base (754M parameters) against ESM-C (600M parameters) on Deep Mutational Scanning (DMS) variant effect prediction across 7 standard assays.
DMS experiments work like this: take a protein, systematically mutate every position to every other amino acid, and measure what happens to function. The resulting dataset tells you, empirically, whether each single mutation helps, hurts, or is neutral. A good PLM should be able to predict this from sequence alone.
For ProGen2, scoring a variant means computing the log-likelihood for both the wildtype and mutant sequences, then taking the difference. A mutation at position 42 changes the log-probability at position 42 and all downstream positions—but positions 1–41 remain identical. The effect ripples forward. Never backward.
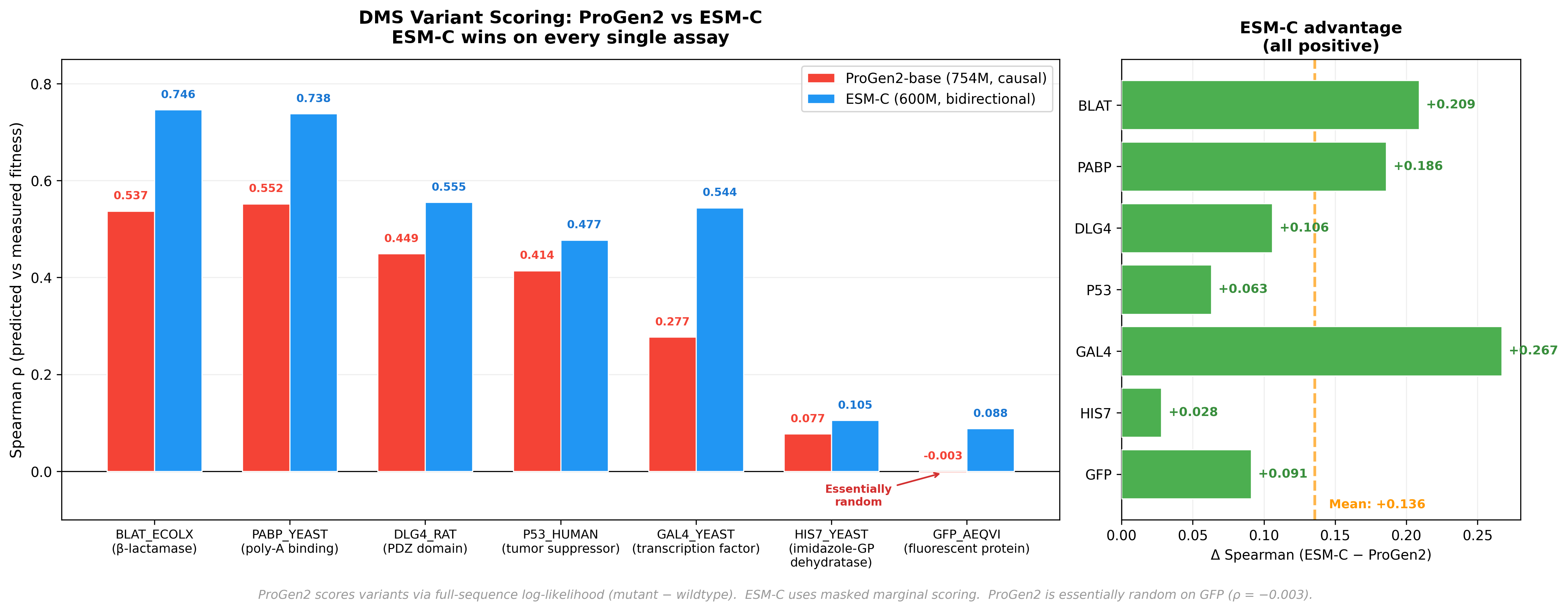
ESM-C outperforms ProGen2 across every assay - a mean Spearman difference of +0.136. The most striking data point: ProGen2 achieves a Spearman correlation of -0.003 on GFP. Essentially random. Now, GFP is arguably the most well-studied protein in all of biology - the field has been staring at it for decades. So this isn’t a case of an obscure protein with sparse training data. It’s a clean signal that something architectural is going on, and it’s worth understanding why.
The answer, we think, is directionality. When ProGen2 scores a mutation, its causal architecture means the effect ripples forward - but never backward. Downstream packing partners and distal contacts that determine whether a mutation is tolerated are invisible to positions upstream of the mutation site. Bidirectional models like ESM-C see both sides. It’s not that ProGen2 is broken- it’s that the autoregressive training objective, so powerful for generation, creates a structural blind spot for variant scoring. The failure mode is specific, it’s explainable, and that’s actually useful.
One important caveat: DMS benchmark performance is heavily influenced by how well a protein family is represented in training data. High performance can reflect memorisation as much as genuine understanding. The bioRxiv preprint “Protein Language Model Fitness Is a Matter of Preference” makes this case convincingly. DMS is our gold standard, and even it is more fragile than we’d like - but that’s a story for another post.
Evals Evals Evals
Everything described above could have been caught earlier. If we had the right evals.
The field currently evaluates PLMs on:
Perplexity — how well does the model predict the next amino acid? A 1D string metric. Tells you nothing about 3D structure, robustness, or compositionality.
DMS fitness prediction — can the model rank single-point mutations? As we showed, heavily influenced by training data distribution, and rewards memorization.
But these new failure modes necessitate different evals and benchmarks: Does this model collapse when given a trivially duplicated sequence? Can it distinguish real 3D structural contacts from decoys? Can it compose two functional constraints it hasn’t seen together? Does it degrade gracefully or fail catastrophically at distribution edges?
Our contact discrimination experiment is, in a sense, a benchmark that didn’t exist before we built it. ProGen2’s AUC of 0.527 would be immediately recognized as failure on any standard ML benchmark. In protein AI, it has gone largely unnoticed.
There’s also a subtler issue: over-optimizing for perplexity can actively hurt. Perplexity rewards the most statistically dense, evolutionarily consensus sequence. But real functional proteins need energetic frustration—sub-optimal packing, dynamic loops, flexible active sites. Pushing perplexity down drives models toward hyper-stable, catalytically inert consensus proteins. You get sequences that look maximally protein-like and do maximally little.
What This Actually Means
We want to be precise about what we’re claiming and what we’re not. As protein design gets easier and easier, knowing the failure modes is what separates successful campaigns v/s spending weeks in the wet-lab.
We’re not saying these models are useless. We use them at Mandrake. They generate high-quality sequences within well-characterized families. Our adapter-based pipeline produces sequences with 98–99% target PFAM identities and meaningful structural diversity. That’s real, and it matters.
We’re saying they are brittle. They work when the problem fits neatly inside the training distribution. They fail—sometimes catastrophically—the moment you step outside that envelope. A trivial duplication trick collapses their predictions. They can’t see 3D contacts. They can’t compose functional constraints. And our benchmarks don’t catch any of this.
We are in the BERT-era of protein AI. These models have learned grammar—which amino acids follow which, what motifs look like, how families cluster in sequence space. They have not learned physics—how proteins fold, how residues interact across 3D space, how functional constraints compose into a working machine.
The next generation of protein AI needs to solve these problems. Whether through better architectures (state-space models and convolutional models show genuine promise), better training objectives (structure-aware pretraining), or better benchmarks that test for the right capabilities—the path forward starts with acknowledging where we actually are.
Not broken. But brittle.
Grammar without physics.
If you liked what you just read, consider subscribing!
 | A guest post by
|
You are absolutely correct. ProGen2 and similar autoregressive protein LMs learn sequence statistics (grammar) but have essentially zero 3D structural awareness. Your key result, ProGen2 AUC of 0.527 for long-range contact discrimination, is statistically indistinguishable from random. The model sees 499 residues of context and still can't tell whether residue 500 is in physical contact with residue 30. More context doesn't help because the model never learned spatial reasoning in the first place.
This is the gap that correlation length eigenspectrum analysis fills. The protein LMs are operating in sequence space, they've learned the 1D statistical structure beautifully. But the physics lives in the eigenspectrum of the electron density: The delocalization index matrix encodes the actual electron-sharing topology, and the eigenspectrum of that matrix captures 3D contact structure by construction because DI(A,B) is large precisely when atoms A and B share significant electron density, regardless of sequence distance.
Your discussion documents three symptoms of the same disease: No 3D contacts: The models never see the electron density that creates contacts. Electron density eigenvalues encode exactly this: the leading eigenvalues of the DI matrix correspond to the dominant delocalization channels, which are the structural backbone of the fold. The resistance of the electron density to perturbation tells you how rigid or flexible those channels are. The model doesn't need to learn contacts from sequence statistics; the physics gives them to you.
Copy bias collapse: This is a pure sequence-space failure. The induction head circuit locks onto token repetition because the model has no physics to override it. An electron density-based representation wouldn't collapse this way; duplicating a sequence doesn't duplicate the eigenspectrum of the electron density, because ρ(r) for ABCABC is not 2×ρ(r) for ABC. The physics breaks the degeneracy that the grammar can't.
DMS scoring failure: Your directionality argument is correct but incomplete. The deeper issue is that variant effect prediction requires knowing how a mutation perturbs the energy landscape, not just the sequence probability. You need to know how the energy redistributes when you mutate a residue; that's what determines whether the mutation is tolerated.'
You say the field needs "structure-aware pretraining" and "better architectures." But you are still thinking within the LM paradigm, i.e learn structure from more data, better objectives. Compute the physics from ρ(r), extract it via electron density to eigenspectrum to stiffness, and use that as the representation. The HK Theorem guarantees this representation is complete, ρ determines everything. No amount of sequence statistics can make that guarantee.