
We don't even design binders!!
We're a gene editor design company in Bangalore. Last month we entered a protein binder design competition as a weekend side-project and ended up winning it. 12000+ designs and only 1 strong binder.

We are a gene editor design company. We work on de novo gene editors to enable a step change upgrade in gene editing and make all those breakthroughs that we’ve been hearing about since years - possible. We do not, as a company, design protein binders.
A few weekends ago we entered the GEM × Adaptyv RBX1 binder design competition. We submitted three different approaches across separate accounts (we just wanted to test our wild appraoches), ran the whole thing as a side project alongside our main gene editing work. This served as a stress-test of our internal AI X biophysical platform on a problem class we don’t normally touch.
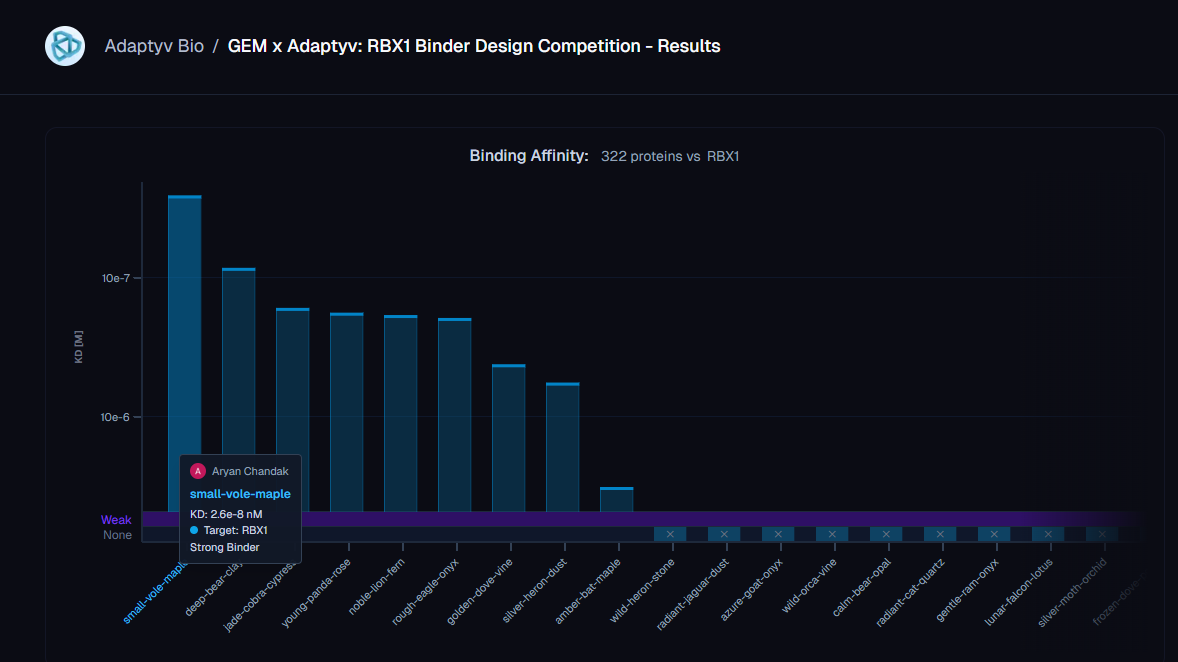
All three of our submissions (21 designs) were selected by Adaptyv for the wet-lab round. 20/21 designs expressed cleanly at the BLI step. One bound - at KD = 26 nM, the only Strong-classified binder in the entire 322-design competition.
The pipeline that produced it is called ORBIT (Oracle-Reseeded Binder design with Interface Targeting). ORBIT surprisingly outperformed teams including Pacesa Lab - the authors of BindCraft, the framework that inspired a range of hallucination based protein design approaches such as Mosaic (And ORBIT itself!!).
All the other teams, focused on designing binders reached medium binding at best. These are serious teams running serious methods. They work on binder design as their primary research. We don’t. But this shows the generalizability of our biophysics approach for enzyme design.
This is the overview. The full technical writeup - methodology, code, ablation logs, design trajectories, candidate structures - is coming in a companion post on The Assay, and we’re releasing everything publicly so the design community can pull it apart, reproduce it, and improve on it. More on that below.
A gorgeous visualization of RBX1 hugging our binder
Why we entered
We’re building an AI X Physics first protein design platform to design de novo gene editing enzymes. As a field, the challenge is how do you search a large search space with as less compute as possible. We’re a fairly young company that’s just started out, and are setting up our wet lab as we speak. This competition was a great way for us to validate some of our hypotheses and see how they stand against actual wet lab results.
The Adaptyv competition was the cleanest version of that benchmark we’d seen. RBX1 partly overlaps with our actual work - metal coordination, partial disorder, multi-domain stability — but is otherwise a different problem class. If our platform could produce a competitive design with weekend-scale effort and three approaches in parallel, that would tell us something. If it couldn’t, that would tell us something too.
We submitted three approaches across separate accounts because we wanted experimental signal across methods, not a single bet. ORBIT was one of the three. The other two were also novel and explored a diverse search space. They get their own writeup. The three-approach result is itself the more interesting story for us internally: most binder design pipelines fail on RBX1 - including two of our own.
Why RBX1 broke most methods
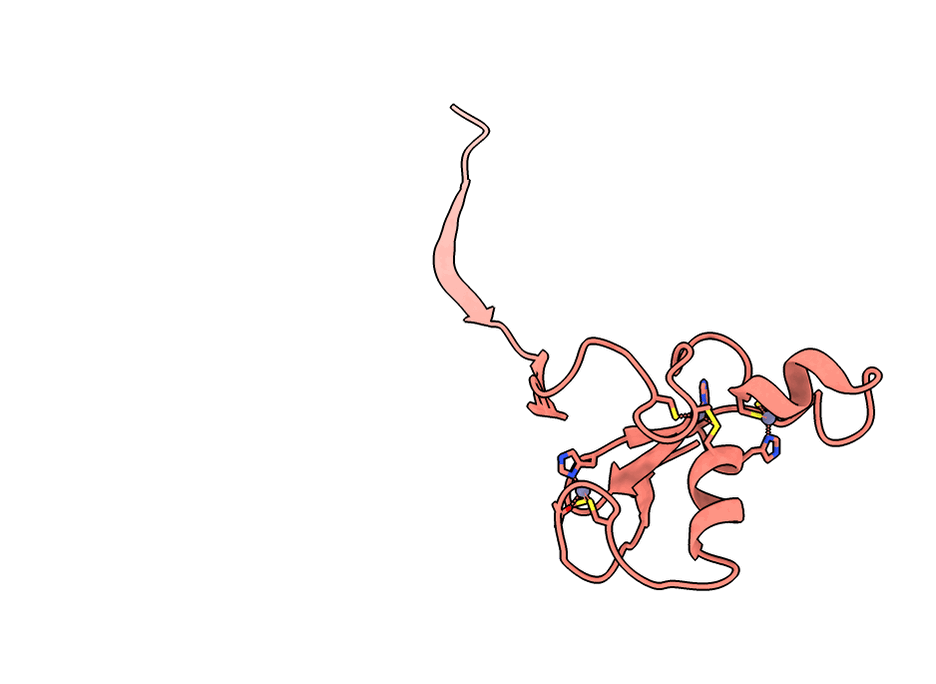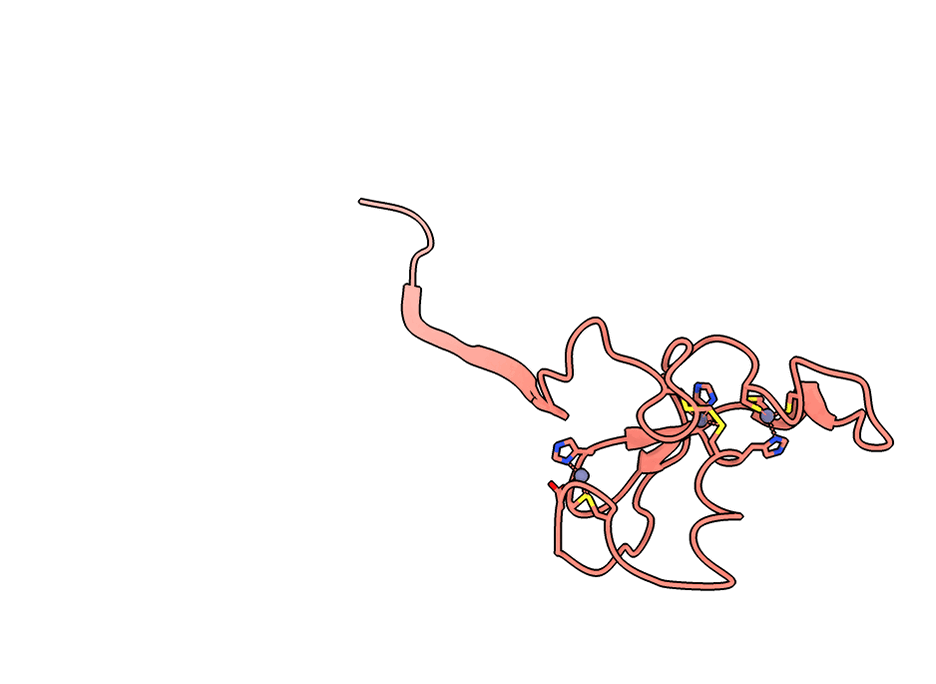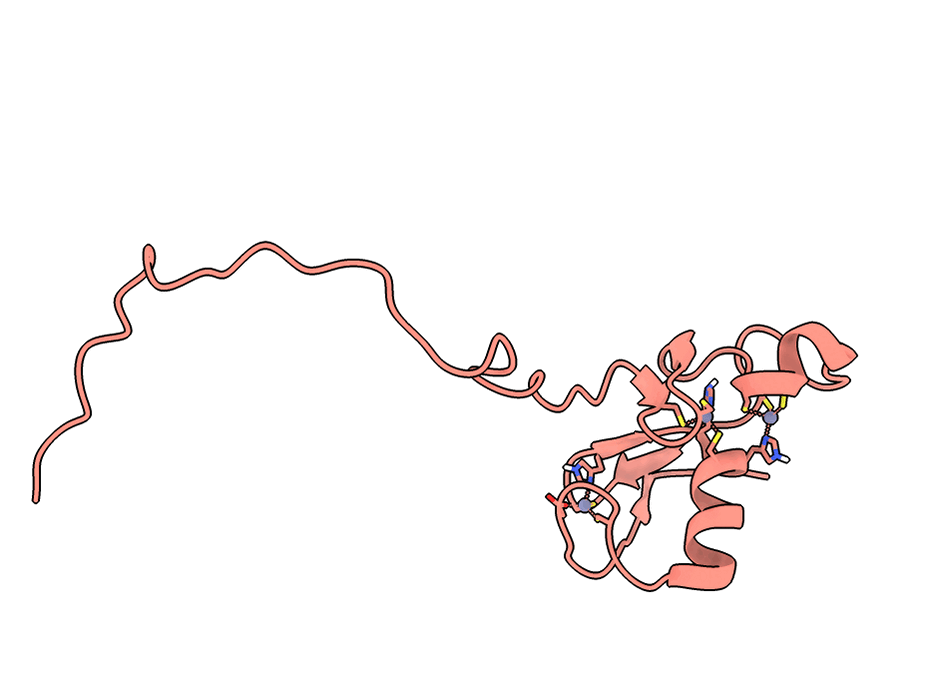
RBX1 is a 108-residue subunit of the Cullin-RING E3 ubiquitin ligase complex. Disrupting its function inhibits CRL-mediated protein degradation, which is therapeutically relevant in cancers where CRL activity is dysregulated. The biology aside, what makes it a brutal computational target is the protein itself.
Three things stack against you. First, an intrinsically disordered N-terminal region that floods standard interface-confidence metrics with noise unrelated to binding. Second, a C-terminal RING-H2 finger that coordinates three Zn²⁺ ions in a cross-brace arrangement - geometry no AI model can infer from sequence alone. Third, no useful evolutionary relatives, so sequence-based methods have nothing to lean on.
The fold-prediction baselines tell you how hard this was: ESMFold pLDDT 0.40 (uninformed), Boltz-2 0.63 (marginal), Protenix without template 0.60. Most de novo binder pipelines start with a structure prediction; if you can’t fold the target cleanly, the entire pipeline downstream inherits the noise.
We used Protenix as the starting point as it gave the best starting prediction as compared to other models. But still, this wasn’t good enough. So, the first useful intervention was the obvious one.
Feed Protenix the 2LGV NMR structure as a template, with three Zn²⁺ ions explicitly placed. pLDDT jumped from 0.60 to 0.86. That single change unlocked everything downstream. For metal-coordinated, constraint-heavy folds, ab initio is a tax you pay for no reason. The template encodes physics - zinc coordination geometry - that no model trained on sequence and general structure can recover. Use it.
ORBIT: three ideas, layered
ORBIT is built on the BindCraft framework that Pacesa Lab developed - backpropagating through a structure prediction model to optimize a sequence PSSM. We use Protenix as the structure prediction model (the JAX version that Escalante Bio built for their Mosaic pipeline) and SolubleMPNN as the inverse folding oracle. Most of what we built sits on top of it.
Search the right regions of design space, and stop searching the wrong ones. Standard differentiable hallucination starts from a uniform PSSM and runs a single long trajectory from there. Most of that compute is spent finding the right neighborhood; only a small fraction is spent refining within it. We changed both halves of that equation.
We turned the optimizer into a multi-stage iterative search. Run a brief hallucination to get a provisional structure. Hand that structure to SolubleMPNN to extract the sequence prior consistent with it. Restart the optimizer from that informed prior. The MPNN sampling temperature is a control knob - low temperatures give a sharp, decisive prior; higher temperatures preserve exploration - and we ran an ablation, then allocated seed volume by what won. The optimizer is no longer doing one long descent from random initialization; it’s doing structured restarts from oracle-informed basins.
We also stopped treating each run as a single function call returning a single answer. A differentiable optimization run is a trajectory through sequence-distribution space. Two consequences. First, the optimizer’s final state isn’t necessarily its best discrete sequence - the argmax projection can be excellent at some intermediate step and worse at the end. We track the best hard sequence along the trajectory and harvest from there. Second, most bad trajectories fail in recognizable ways early. A small classifier trained on intermediate trajectory features catches failures at step 60 of a 165-step run with 97.7% accuracy and zero false negatives at the conservative threshold. Together: dead runs stop consuming budget, live ones get harvested for their best discrete output rather than their final one, and high-variance branches - the ones with the highest peaks - become affordable to scale.
A force-field for the interface. Most binder design pipelines treat the target surface as uniform - bind anywhere. Some allow weighted contact targeting in distogram space, which biases where contacts form but doesn’t shape how the binder gets pulled there. We built a gravitational binding loss that operates in coordinate space directly: high-value target residues exert a continuous attractive field on the binder, with the strength of the pull modulated by per-residue importance. The framework extends naturally to repulsive zones and exclusion regions. RBX1 was the first target we built it for; the abstraction is general.
A programmable biophysical layer. We added a regularization layer that operates on the soft PSSM during optimization - soft pressure toward foldability margin, compositional balance, controlled net charge, hydrophobicity profile, manufacturability. The structural model is no longer the only judge of what the optimizer should care about. The optimizer feels biophysical intent throughout the trajectory, not just as a downstream filter. This is the part of the system that’s genuinely platform-shaped: switch the regularizers, and the same optimization machinery steers toward a different biochemical profile. The work it did on RBX1 is one configuration of a more general capability.
Apart from these three key research ideas above there’s a longer engineering stack underneath that we use day-to-day in our pipeline such as: Gumbel-Softmax sampling, slow PSSM sharpening, cosine-decayed steering schedules, noise-robust interface scoring, a few other things.
The full technical writeup is going up on The Assay in the detailed post, soon.
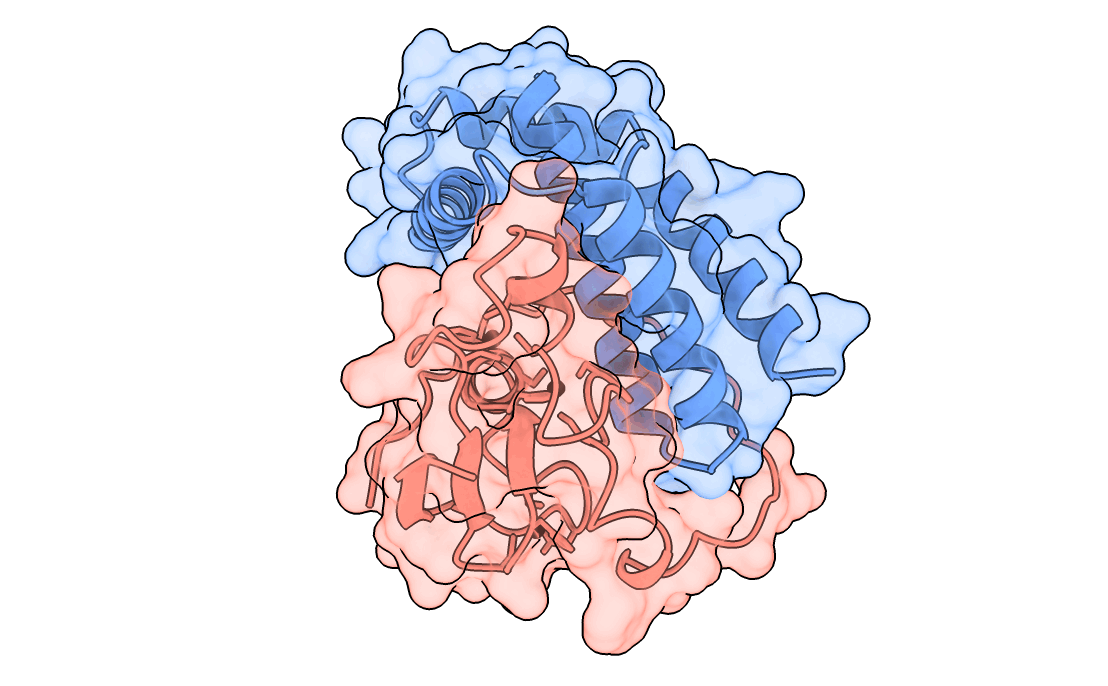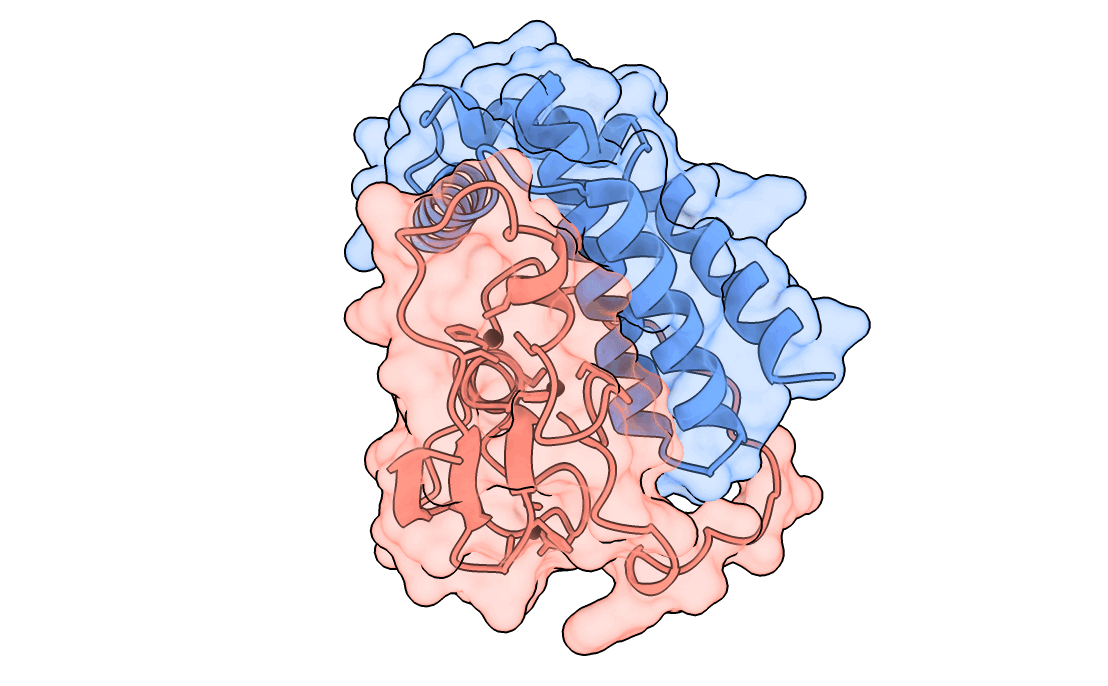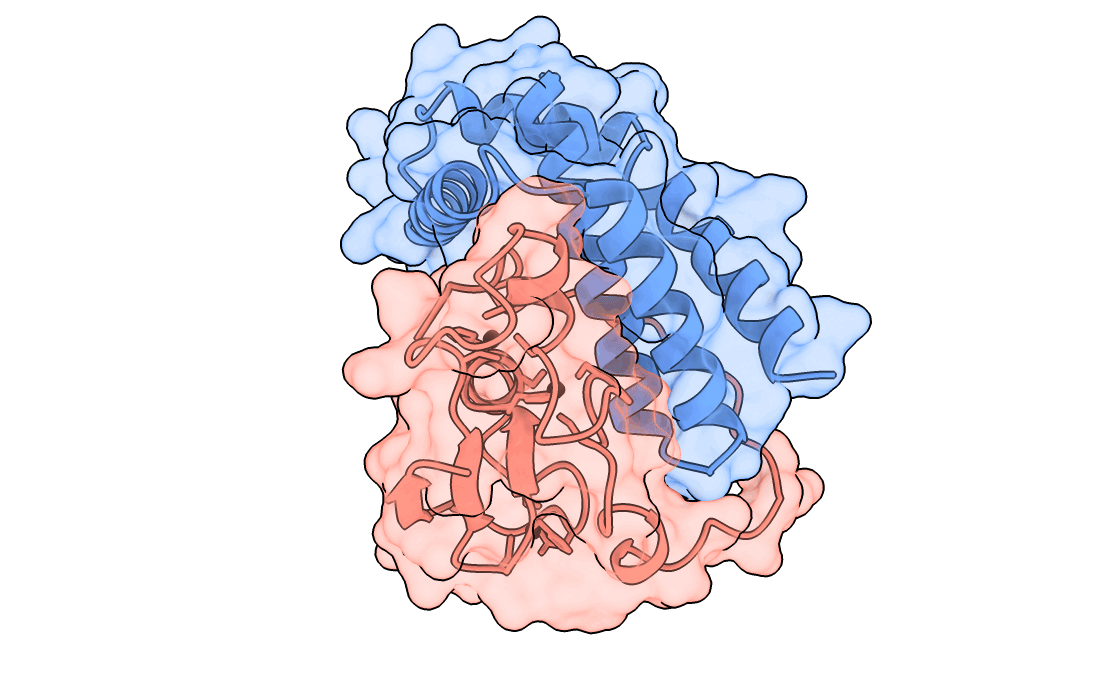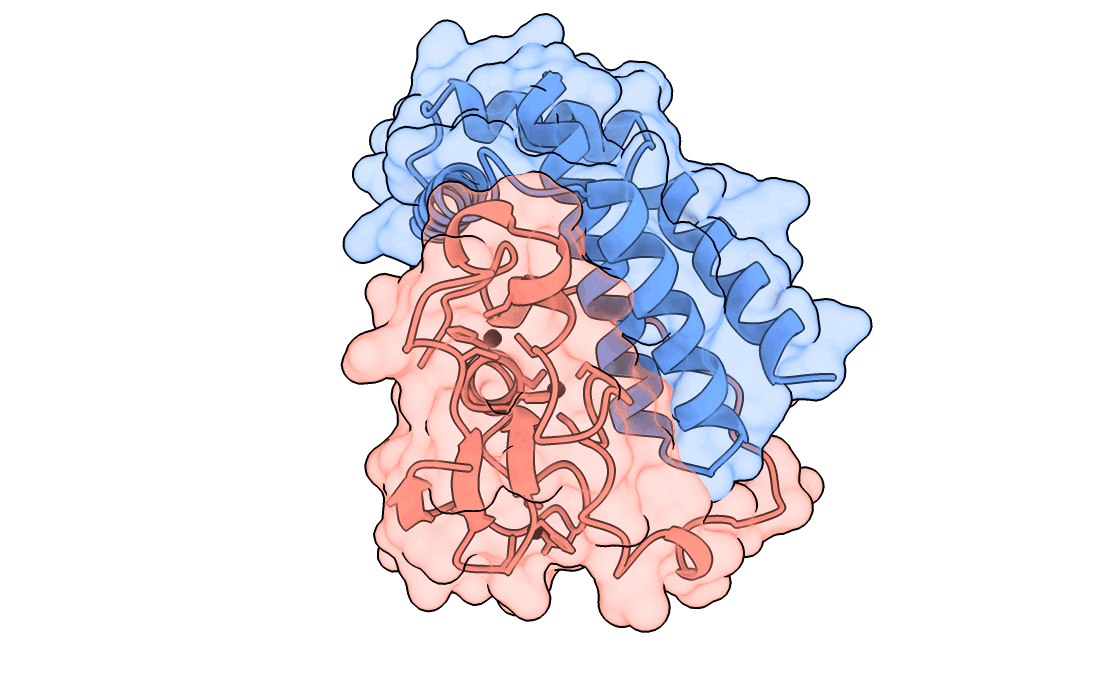
One important point to note – We also saw that the past Adaptyv data showed model metrics such as ipTM, pLDDT, have significantly less correlation with actual binder performance, so every design which scored high on our metrics had to pass a strict ‘vibe check’ by our protein designer – Surabhi, for it to make it to the final list.
The result
Our winning binder is a 100-residue, 11.8 kDa de novo binder. It expressed cleanly, folded independently as a stable monomer (pLDDT 0.975), and bound RBX1 at K_D = 2.6 × 10⁻⁸ M (26 nM) by bio-layer interferometry.
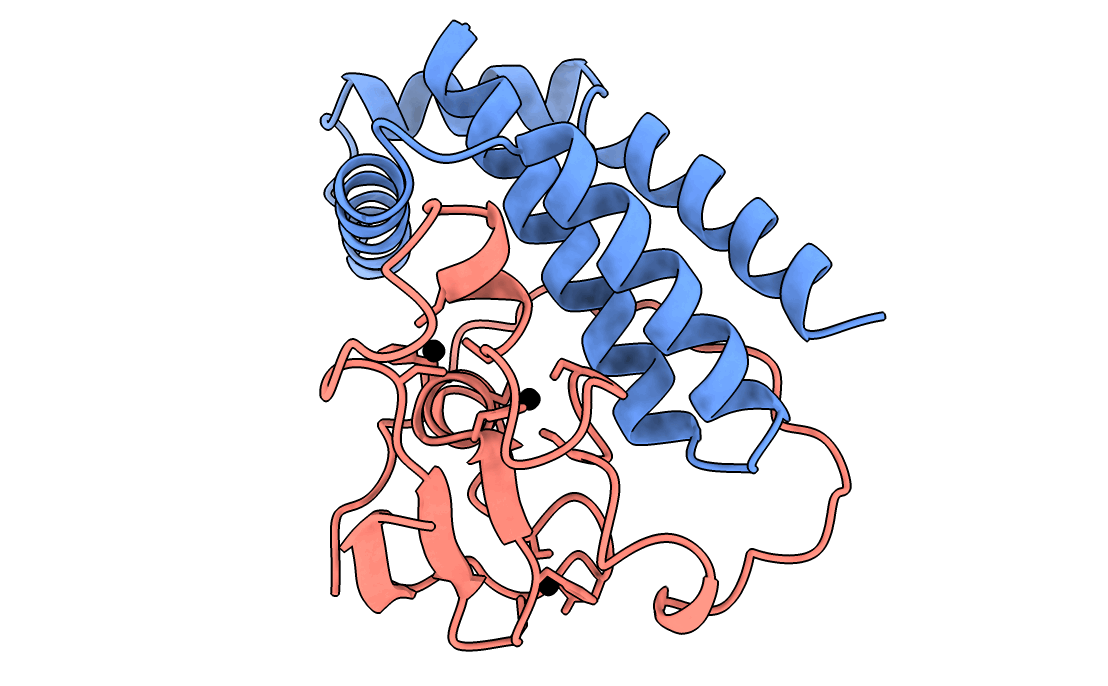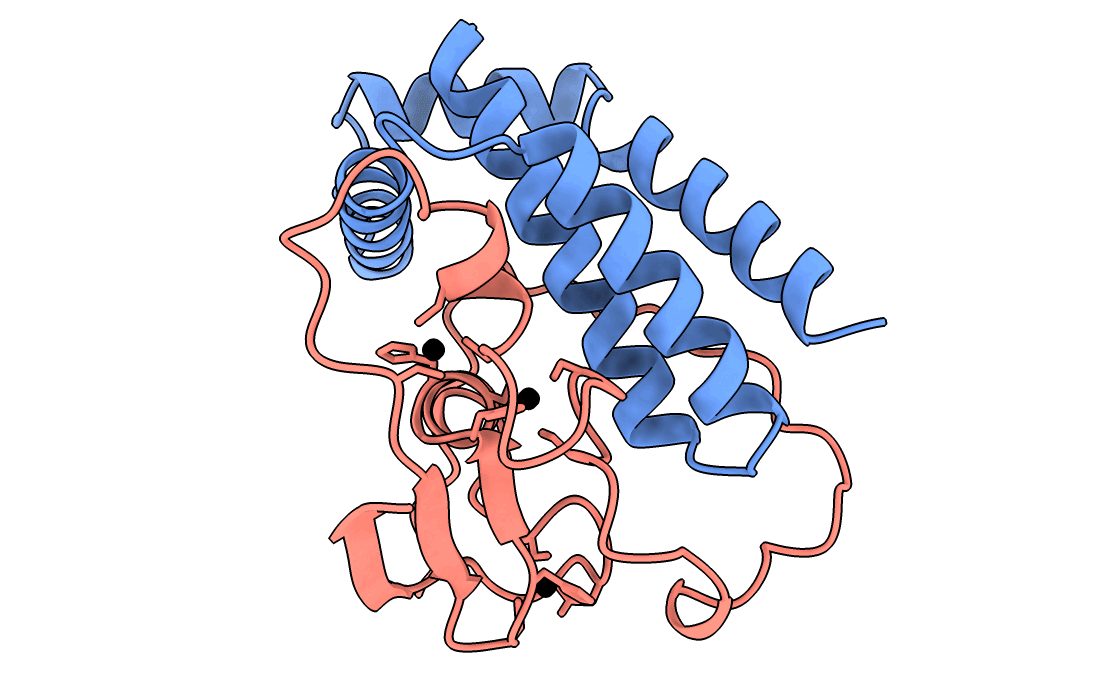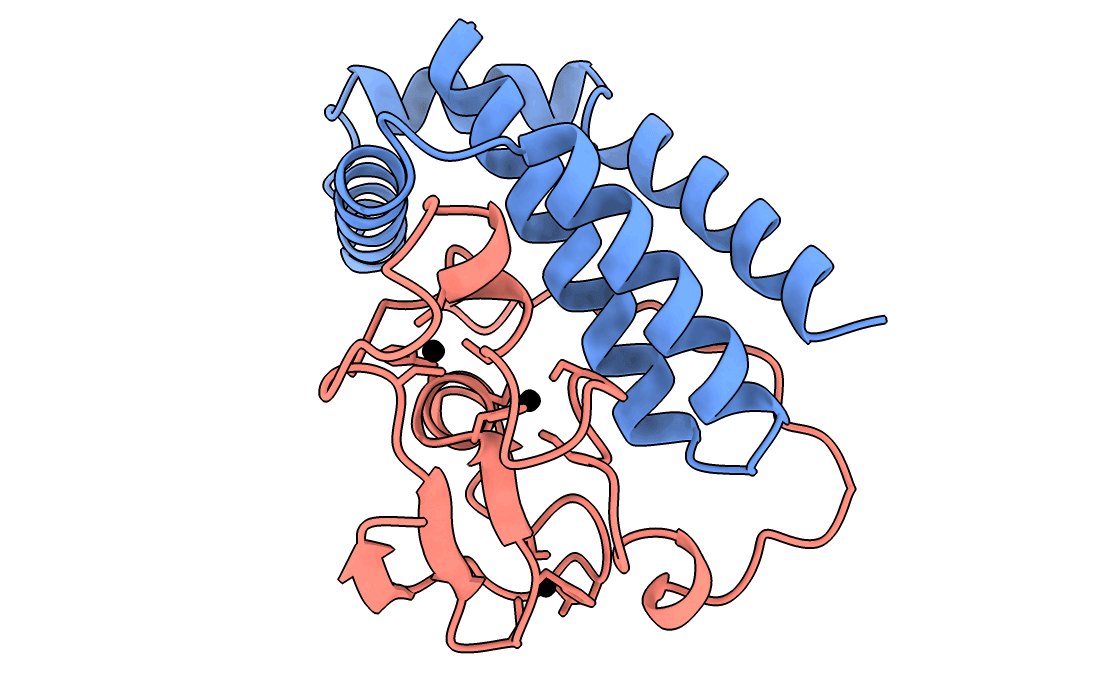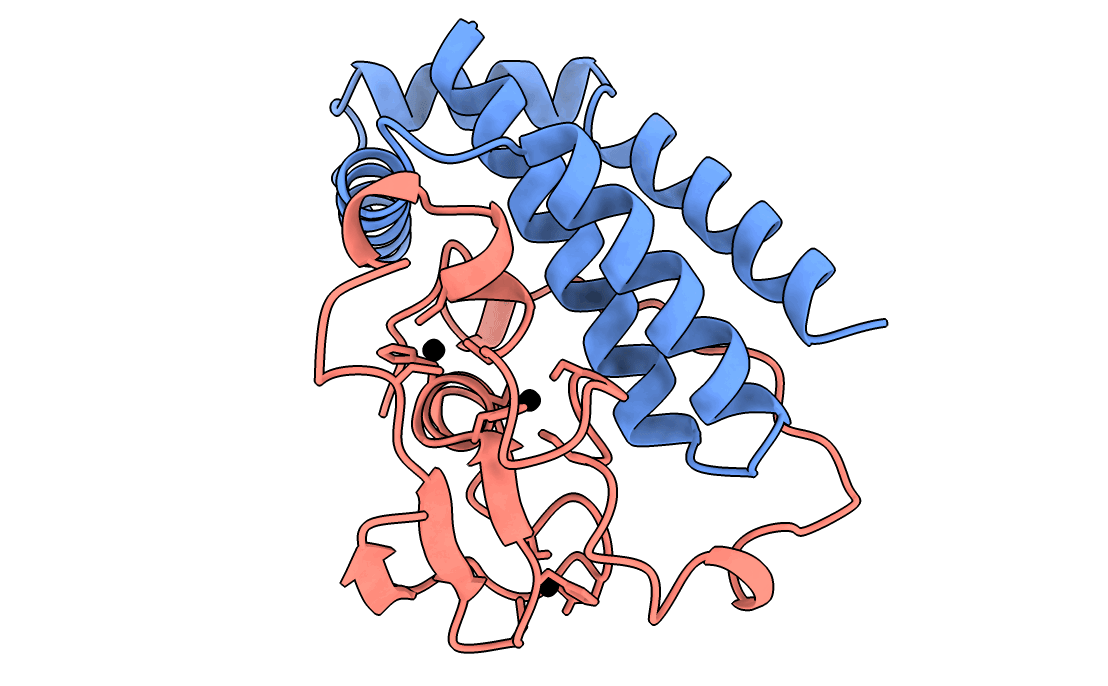
Adaptyv’s classification: Strong. The only one in that category across all 322 designs in the competition.
As Martin Pacesa says - Helices are all you need :p
The next-best binder came in at 85 nM - a 3.3× gap in absolute affinity, but within the same order of magnitude. The cleaner reading is the categorical one: ours was the only design Adaptyv classified as Strong, the next eight binders were Medium (170 nM to 5.7 × 10⁻⁷ M), and 313 of 322 designs did not bind the target at all. That hit-rate is the more honest summary of where the field is on this kind of target - partially-disordered, metal-coordinated, no useful structural homologs. Designing binders for proteins that don’t sit still is genuinely unsolved.
Which is also the framing we want to put on the result. One Strong binder on RBX1 is not “binder design for IDPs is solved.” It’s a single data point that the right integration choices - template guidance, structure-informed reseeding, weighted epitope steering, multi-model gating - can find affinity in design space that earlier configurations missed. That space is large. Most of it is still unmapped.
What’s coming: open release
We engineered a bunch of things across the pipeline. Code, ablations, and a detailed end-to-end writeup of exactly what we did and why are coming in a follow-up post on The Assay - subscribe there if you want it in your inbox when it lands.
The competition’s full dataset is already open under ODC-ODbL on Proteinbase; ORBIT will sit alongside it for anyone to pull apart, reproduce, and improve on.
If you have built or are building a binder design pipeline and want to compare notes - or if you find something in our methodology you’d push back on - please. That’s the point.
What’s next
The hardest protein design problems we work on at Mandrake aren’t binders - they’re enzymes whose function depends on dynamic conformational states no current structure prediction model handles cleanly. RBX1 was a useful stress-test, but the open problem we actually care about is generating designs that get state-dependent function right on the first try. That’s where the next wave of methods has to land. We’re working on it.
Thanks to the GEM Workshop and Adaptyv Bio for organizing a competition that takes hard targets seriously, and to Adaptyv’s experimental team for fast, careful BLI work. Particular thanks to Pacesa Lab for BindCraft, and Escalante Bio for Mosaic which inspired the differentiable hallucination core of ORBIT.
If you read this and have something you’d push back on - or if you’re an AI Researcher, physicist or protein engineer interested in this kind of work - we are hiring.
Reach us at ai@mandrake.bio
Mandrake Bio: foundational protein engineering for next-generation gene editors. Bengaluru.
If you liked this post, consider sharing it with others in your network!
Congratulations Team Mandrake
Congrats!